

What Are The Sizes Of A Molecule, Bacteria, Animal Cell, Virus, And Plant Cell?
A typical animal cell is 10–20 μm in bore, which is nigh i-fifth the size of the smallest particle visible to the naked eye. Information technology was not until skillful light microscopes became available in the early function of the nineteenth century that all plant and beast tissues were discovered to be aggregates of private cells. This discovery, proposed as the prison cell doctrine past Schleiden and Schwann in 1838, marks the formal birth of jail cell biology.
Animal cells are not only tiny, they are also colorless and translucent. Consequently, the discovery of their primary internal features depended on the evolution, in the latter function of the nineteenth century, of a diversity of stains that provided sufficient dissimilarity to brand those features visible. Similarly, the introduction of the far more than powerful electron microscope in the early 1940s required the development of new techniques for preserving and staining cells earlier the full complexities of their internal fine structure could brainstorm to emerge. To this twenty-four hours, microscopy depends as much on techniques for preparing the specimen equally on the performance of the microscope itself. In the discussions that follow, nosotros therefore consider both instruments and specimen training, beginning with the light microscope.
Figure 9-1 shows a series of images illustrating an imaginary progression from a thumb to a cluster of atoms. Each successive prototype represents a tenfold increment in magnification. The naked centre could encounter features in the first ii panels, the resolution of the light microscope would extend to near the fourth console, and the electron microscope to virtually the 7th console. Some of the landmarks in the development of light microscopy are outlined in Tabular array nine-1. Figure 9-2 shows the sizes of various cellular and subcellular structures and the ranges of size that dissimilar types of microscopes tin visualize.

Effigy nine-one
A sense of calibration between living cells and atoms. Each diagram shows an image magnified by a factor of ten in an imaginary progression from a thumb, through skin cells, to a ribosome, to a cluster of atoms forming part of one of the many protein molecules (more...)
Table nine-1
Some Of import Discoveries in the History of Lite Microscopy.

Figure nine-two
Resolving power. Sizes of cells and their components are drawn on a logarithmic calibration, indicating the range of objects that can be readily resolved past the naked eye and in the lite and electron microscopes. The post-obit units of length are normally (more...)
The Calorie-free Microscope Can Resolve Details 0.two μm Apart
In general, a given blazon of radiations cannot exist used to probe structural details much smaller than its own wavelength. This is a key limitation of all microscopes. The ultimate limit to the resolution of a light microscope is therefore set by the wavelength of visible lite, which ranges from about 0.4 μm (for violet) to 0.7 μm (for deep scarlet). In applied terms, bacteria and mitochondria, which are about 500 nm (0.five μm) wide, are mostly the smallest objects whose shape can be conspicuously discerned in the light microscope; details smaller than this are obscured by effects resulting from the wave nature of lite. To empathise why this occurs, we must follow what happens to a beam of calorie-free waves equally it passes through the lenses of a microscope (Figure ix-3).
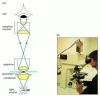
Figure 9-iii
A light microscope. (A) Diagram showing the light path in a compound microscope. Light is focused on the specimen by lenses in the condensor. A combination of objective lenses and eyepiece lenses are arranged to focus an image of the illuminated specimen (more...)
Because of its wave nature, light does not follow exactly the idealized straight ray paths predicted by geometrical eyes. Instead, low-cal waves travel through an optical system by a variety of slightly different routes, then that they interfere with one another and cause optical diffraction effects. If two trains of waves reaching the same betoken past unlike paths are precisely in stage, with crest matching crest and trough matching trough, they will reinforce each other so as to increase brightness. In contrast, if the trains of waves are out of phase, they volition interfere with each other in such a way equally to cancel each other partly or entirely (Figure 9-iv). The interaction of light with an object changes the phase relationships of the low-cal waves in a manner that produces circuitous interference effects. At loftier magnification, for example, the shadow of a straight edge that is evenly illuminated with light of compatible wavelength appears as a set of parallel lines, whereas that of a circular spot appears as a set of concentric rings (Figure 9-5). For the same reason, a unmarried point seen through a microscope appears as a blurred disc, and 2 point objects close together give overlapping images and may merge into one. No amount of refinement of the lenses can overcome this limitation imposed by the wavelike nature of light.

Figure 9-iv
Interference between light waves. When 2 light waves combine in phase, the aamplitude of the resultant wave is larger and the brightness is increased. Two low-cal waves that are out of phase cancel each other partly and produce a moving ridge whose amplitude, (more than...)

Effigy 9-5
Border furnishings. The interference furnishings observed at high magnification when calorie-free passes the edges of a solid object placed between the light source and the observer are shown here.
The limiting separation at which two objects can nonetheless be seen equally singled-out—the so-called limit of resolution—depends on both the wavelength of the light and the numerical aperture of the lens arrangement used. This latter quantity is a measure of the width of the entry pupil of the microscope, scaled according to its altitude from the object; the wider the microscope opens its middle, and so to speak, the more sharply it can meet (Figure ix-vi). Under the best conditions, with violet light (wavelength = 0.iv μm) and a numerical aperture of ane.4, a limit of resolution of just under 0.2 μm can theoretically be obtained in the low-cal microscope. This resolution was accomplished by microscope makers at the stop of the nineteenth century and is but rarely matched in gimmicky, factory-produced microscopes. Although it is possible to overstate an epitome as much as 1 wants—for example, past projecting information technology onto a screen—it is never possible to resolve two objects in the low-cal microscope that are separated by less than about 0.2 μm; they will appear equally a single object.

Figure nine-6
Numerical aperture. The path of light rays passing through a transparent specimen in a microscope illustrate the concept of numerical aperture and its relation to the limit of resolution.
We see next how interference and diffraction tin can exist exploited to study unstained cells in the living country. Subsequently we discuss how permanent preparations of cells are made for viewing in the calorie-free microscope and how chemical stains are used to enhance the visibility of the cell structures in such preparations.
Living Cells Are Seen Clearly in a Phase-Contrast or a Differential-Interference-Contrast Microscope
The possibility that some components of the cell may be lost or distorted during specimen preparation has e'er challenged microscopists. The only certain manner to avert the problem is to examine cells while they are alive, without fixing or freezing. For this purpose, low-cal microscopes with special optical systems are especially useful.
When light passes through a living cell, the phase of the light moving ridge is changed according to the prison cell's refractive index: light passing through a relatively thick or dense function of the jail cell, such equally the nucleus, is retarded; its phase, consequently, is shifted relative to light that has passed through an adjacent thinner region of the cytoplasm. The phase-dissimilarity microscope and, in a more complex mode, the differential-interference-contrast microscope, exploit the interference effects produced when these two sets of waves recombine, thereby creating an prototype of the jail cell's construction (Figure 9-7). Both types of calorie-free microscopy are widely used to visualize living cells.

Figure 9-7
Two ways to obtain contrast in light microscopy. (A) The stained portions of the cell reduce the amplitude of light waves of particular wavelengths passing through them. A colored image of the cell is thereby obtained that is visible in the ordinary style. (more...)
A simpler fashion to see some of the features of a living cell is to observe the light that is scattered past its diverse components. In the dark-field microscope, the illuminating rays of light are directed from the side and so that but scattered light enters the microscope lenses. Consequently, the prison cell appears every bit a vivid object against a dark groundwork. With a normal bright-field microscope, the image is obtained by the simple transmission of light through a cell in culture. Images of the aforementioned prison cell obtained by 4 kinds of low-cal microscopy are shown in Figure ix-eight.

Effigy 9-8
Four types of light microscopy. Four images are shown of the aforementioned fibroblast cell in culture. All four types of images tin exist obtained with virtually modern microscopes by interchanging optical components. (A) Vivid-field microscopy. (B) Phase-contrast microscopy. (more...)
Phase-dissimilarity, differential-interference-contrast, and night-field micros-copy make it possible to watch the movements involved in such processes as mitosis and jail cell migration. Since many cellular motions are too ho-hum to exist seen in real fourth dimension, it is often helpful to take time-lapse motion pictures or video recordings. Here, successive frames separated by a short time delay are recorded, and so that when the resulting flick series or videotape is played at normal speed, events appear profoundly speeded up.
Images Can Be Enhanced and Analyzed past Electronic Techniques
In contempo years electronic imaging systems and the associated applied science of image processing have had a major impact on light microscopy. They have enabled certain practical limitations of microscopes (due to imperfections in the optical system) to be largely overcome. They have also circumvented two cardinal limitations of the human eye: the eye cannot see well in extremely dim light, and it cannot perceive modest differences in light intensity against a bright background. The showtime limitation can exist overcome by attaching highly sensitive video cameras (the kind used in night surveillance) to a microscope. It is and then possible to find cells for long periods at very low light levels, thereby avoiding the dissentious furnishings of prolonged bright light (and heat). Such low-light cameras are particularly important for viewing fluorescent molecules in living cells, as explained below.
Because images produced by video cameras are in electronic form, they can exist readily digitized, fed to a computer, and processed in various ways to extract latent data. Such image processing makes information technology possible to compensate for diverse optical faults in microscopes to attain the theoretical limit of resolution. Moreover, by electronic image processing, contrast can be greatly enhanced so that the eye's limitations in detecting minor differences in low-cal intensity are overcome. Although this processing too enhances the effects of random background irregularities in the optical system, such defects can exist removed by electronically subtracting an prototype of a blank area of the field. Small transparent objects that were previously incommunicable to distinguish from the background and so go visible.
The high contrast attainable by reckoner-assisted differential-interference-contrast microscopy makes information technology possible to see even very small objects such as single microtubules (Effigy nine-9), which have a diameter of 0.025 μm, less than one-10th the wavelength of lite. Individual microtubules tin also be seen in a fluorescence microscope if they are fluorescently labeled (see Figure ix-15). In both cases, however, the unavoidable diffraction effects badly blur the image and then that the microtubules appear at to the lowest degree 0.ii μm wide, making it incommunicable to distinguish a single microtubule from a package of several microtubules.

Figure 9-ix
Image processing. (A) Unstained microtubules are shown here in an unprocessed digital image, captured using differential-interference-contrast microscopy. (B) The image has now been processed, offset by digitally subtracting the unevenly illuminated background, (more...)
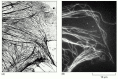
Figure nine-15
Immunofluorescence. (A) A transmission electron micrograph of the periphery of a cultured epithelial jail cell showing the distribution of microtubules and other filaments. (B) The same expanse stained with fluorescent antibodies against tubulin, the protein (more...)
Tissues Are Usually Fixed and Sectioned for Microscopy
To make a permanent preparation that can exist stained and viewed at leisure in the microscope, i first must care for cells with a fixative so as to immobilize, kill, and preserve them. In chemical terms, fixation makes cells permeable to staining reagents and cantankerous-links their macromolecules then that they are stabilized and locked in position. Some of the earliest fixation procedures involved immersion in acids or in organic solvents, such as alcohol. Current procedures normally include treatment with reactive aldehydes, particularly formaldehyde and glutaraldehyde, which class covalent bonds with the gratuitous amino groups of proteins and thereby cross-link next protein molecules.
Virtually tissue samples are too thick for their individual cells to exist examined straight at high resolution. After fixation, therefore, the tissues are usually cut into very sparse slices, or sections, with a microtome, a machine with a abrupt blade that operates like a meat slicer (Figure 9-10). The sections (typically one–10 μm thick) are then laid flat on the surface of a glass microscope slide.

Figure 9-x
Making tissue sections. This illustration shows how an embedded tissue is sectioned with a microtome in preparation for exam in the light microscope.
Because tissues are generally soft and fragile, even after fixation, they demand to exist embedded in a supporting medium before sectioning. The usual embedding media are waxes or resins. In liquid grade these media both permeate and surround the stock-still tissue; they can and then exist hardened (by cooling or by polymerization) to class a solid block, which is readily sectioned by the microtome.
In that location is a serious danger that any treatment used for fixation and embedding may change the construction of the cell or its constituent molecules in undesirable means. Rapid freezing provides an alternative method of training that to some extent avoids this trouble by eliminating the need for fixation and embedding. The frozen tissue can be cutting directly with a special microtome that is maintained in a cold bedroom. Although frozen sections produced in this way avert some artifacts, they endure from others: the native structures of individual molecules such as proteins are well preserved, simply the fine construction of the cell is oft disrupted past water ice crystals.
Once sections have been cutting, by whatever method, the next step is commonly to stain them.
Different Components of the Cell Can Be Selectively Stained
There is little in the contents of nigh cells (which are seventy% water by weight) to impede the passage of light rays. Thus, most cells in their natural country, even if fixed and sectioned, are well-nigh invisible in an ordinary low-cal microscope. One fashion to make them visible is to stain them with dyes.
In the early nineteenth century, the demand for dyes to stain textiles led to a fertile period for organic chemistry. Some of the dyes were found to stain biological tissues and, unexpectedly, often showed a preference for detail parts of the jail cell—the nucleus or mitochondria, for example—making these internal structures clearly visible. Today a rich variety of organic dyes is available, with such colorful names as Malachite green, Sudan blackness, and Coomassie blueish, each of which has some specific affinity for item subcellular components. The dye hematoxylin, for example, has an affinity for negatively charged molecules and therefore reveals the distribution of Dna, RNA, and acidic proteins in a cell (Figure 9-11). The chemical basis for the specificity of many dyes, however, is not known.
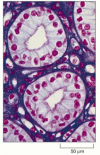
Figure nine-11
A stained tissue section. . This section of cells in the urine-collecting ducts of the kidney was stained with a combination of dyes, hematoxylin and eosin, commonly used in histology. Each duct is made of closely packed cells (with nuclei stained crimson) (more...)
The relative lack of specificity of these dyes at the molecular level has stimulated the design of more than rational and selective staining procedures and, in particular, of methods that reveal specific proteins or other macromolecules in cells. It is a problem, however, to attain adequate sensitivity for this purpose. Since relatively few copies of most macromolecules are nowadays in any given cell, ane or ii molecules of stain bound to each macromolecule are ofttimes invisible. One fashion to solve this problem is to increment the number of stain molecules associated with a single macromolecule. Thus, some enzymes can be located in cells through their catalytic activeness: when supplied with appropriate substrate molecules, each enzyme molecule generates many molecules of a localized, visible reaction production. An alternative and much more than generally applicative approach to the problem of sensitivity depends on using dyes that are fluorescent, as we explicate next.
Specific Molecules Can Be Located in Cells by Fluorescence Microscopy
Fluorescent molecules blot light at one wavelength and emit it at another, longer wavelength. If such a chemical compound is illuminated at its absorbing wavelength and then viewed through a filter that allows only lite of the emitted wavelength to pass, it is seen to glow confronting a dark groundwork. Because the groundwork is dark, even a minute amount of the glowing fluorescent dye can exist detected. The same number of molecules of an ordinary stain viewed conventionally would be practically invisible because they would give only the faintest tinge of colour to the light transmitted through this stained role of the specimen.
The fluorescent dyes used for staining cells are detected by a fluorescence microscope. This microscope is similar to an ordinary light microscope except that the illuminating light, from a very powerful source, is passed through two sets of filters—one to filter the light before information technology reaches the specimen and ane to filter the lite obtained from the specimen. The offset filter is selected so that it passes only the wavelengths that excite the particular fluorescent dye, while the 2d filter blocks out this light and passes only those wavelengths emitted when the dye fluoresces (Figure 9-12).

Effigy 9-12
The optical organisation of a fluorescence microscope. A filter set consists of 2 barrier filters (1 and iii) and a dichroic (beam-splitting) mirror (2). In this example, the filter set for detection of the fluorescent molecule fluorescein is shown. High-numerical-aperture (more...)
Fluorescence microscopy is nearly often used to observe specific proteins or other molecules in cells and tissues. A very powerful and widely used technique is to couple fluorescent dyes to antibody molecules, which then serve as highly specific and versatile staining reagents that bind selectively to the particular macromolecules they recognize in cells or in the extracellular matrix. Two fluorescent dyes that have been ordinarily used for this purpose are fluorescein, which emits an intense dark-green fluorescence when excited with blue light, and rhodamine, which emits a deep scarlet fluorescence when excited with green-yellowish light (Effigy 9-xiii). By coupling one antibody to fluorescein and another to rhodamine, the distributions of different molecules can be compared in the aforementioned jail cell; the two molecules are visualized separately in the microscope by switching back and forth between ii sets of filters, each specific for i dye. Every bit shown in Figure 9-14, three fluorescent dyes can be used in the same way to distinguish between three types of molecules in the same cell. Many newer fluorescent dyes, such as Cy3, Cy5, and the Alexa dyes, have been specifically developed for fluorescence microscopy (see Figure 9-xiii).

Figure 9-13
Fluorescent dyes. The maximum excitation and emission wavelengths of several ordinarily used fluorescent dyes are shown in relation to the corresponding colours of the spectrum. The photon emitted by a dye molecule is necessarily of lower energy (longer (more...)

Figure 9-14
Multiple-fluorescent-probe microscopy. In this composite micrograph of a cell in mitosis, 3 dissimilar fluorescent probes have been used to stain 3 different cellular components. The spindle microtubules are revealed with a greenish fluorescent antibody, (more than...)
Of import methods, discussed afterwards in the affiliate, enable fluorescence microscopy to be used to monitor changes in the concentration and location of specific molecules within living cells (see p. 574).
Antibodies Can Be Used to Observe Specific Molecules
Antibodies are proteins produced by the vertebrate allowed system as a defense against infection (discussed in Affiliate 24). They are unique amidst proteins because they are made in billions of dissimilar forms, each with a different binding site that recognizes a specific target molecule (or antigen). The precise antigen specificity of antibodies makes them powerful tools for the cell biologist. When labeled with fluorescent dyes, they are invaluable for locating specific molecules in cells by fluorescence microscopy (Figure 9-15); labeled with electron-dumbo particles such as colloidal gold spheres, they are used for similar purposes in the electron microscope (discussed below).
The sensitivity of antibodies every bit probes for detecting and assaying specific molecules in cells and tissues is frequently enhanced past chemical methods that amplify the indicate. For case, although a marker molecule such equally a fluorescent dye can be linked directly to an antibiotic used for specific recognition—the chief antibiotic—a stronger point is achieved by using an unlabeled primary antibody then detecting it with a group of labeled secondary antibodies that bind to it (Figure 9-16).

Figure 9-16
Indirect immuno-cytochemistry. This detection method is very sensitive considering the primary antibody is itself recognized past many molecules of the secondary antibody. The secondary antibody is covalently coupled to a marker molecule that makes it readily (more...)
The well-nigh sensitive amplification methods utilize an enzyme as a mark molecule attached to the secondary antibody. The enzyme element of group i phosphatase, for example, in the presence of appropriate chemicals, produces inorganic phosphate and leads to the local formation of a colored precipitate. This reveals the location of the secondary antibody that is coupled to the enzyme and hence the location of the antibiotic-antigen complex to which the secondary antibody is bound. Since each enzyme molecule acts catalytically to generate many thousands of molecules of production, even tiny amounts of antigen tin be detected. An enzyme-linked immunosorbent analysis (ELISA) based on this principle is frequently used in medicine every bit a sensitive test—for pregnancy or for various types of infections, for example. Although the enzyme amplification makes enzyme-linked methods very sensitive, diffusion of the colored precipitate abroad from the enzyme means that the spatial resolution of this method for microscopy may be limited, and fluorescent labels are commonly used for the virtually precise optical localization.
Antibodies are made most simply by injecting a sample of the antigen several times into an animal such as a rabbit or a goat and then collecting the antibody-rich serum. This antiserum contains a heterogeneous mixture of antibodies, each produced by a different antibody-secreting cell (a B lymphocyte). The dissimilar antibodies recognize various parts of the antigen molecule (called an antigenic determinant, or epitope), equally well as impurities in the antigen training. The specificity of an antiserum for a particular antigen can sometimes be sharpened past removing the unwanted antibody molecules that bind to other molecules; an antiserum produced against protein X, for instance, tin be passed through an affinity column of antigens Y and Z to remove any contaminating anti-Y and anti-Z antibodies. Even and so, the heterogeneity of such antisera sometimes limits their usefulness. This problem is largely overcome by the employ of monoclonal antibodies (see Figure viii-half dozen). However, monoclonal antibodies can also have problems. Since they are single antibody protein species, they show almost perfect specificity for a unmarried site or epitope on the antigen, only the accessibility of the epitope, and thus the usefulness of the antibody, may depend on the specimen training. For example, some monoclonal antibodies volition react only with unfixed antigens, others only later the utilise of particular fixatives, and still others only with proteins denatured on SDS polyacrylamide gels, and non with the proteins in their native conformation.
Imaging of Complex Iii-dimensional Objects Is Possible with the Optical Microscope
For ordinary light microscopy, as we have seen, a tissue has to be sliced into thin sections to be examined; the thinner the section, the crisper the paradigm. In the procedure of sectioning, information near the third dimension is lost. How, so, can one become a moving picture of the three-dimensional architecture of a cell or tissue, and how tin can one view the microscopic structure of a specimen that, for one reason or another, cannot showtime be sliced into sections? Although an optical microscope is focused on a particular focal plane within circuitous three-dimensional specimens, all the other parts of the specimen higher up and beneath the plane of focus are besides illuminated, and the light originating from these regions contributes to the prototype as "out-of-focus" blur. This can make information technology very hard to translate the image in item, and can lead to fine prototype structure being obscured by the out-of-focus calorie-free.
Two approaches have been developed to solve this problem: one is computational, the other is optical. These 3-dimensional microscopic imaging methods get in possible to focus on a chosen airplane in a thick specimen while rejecting the light that comes from out-of-focus regions to a higher place and below that airplane. Thus 1 sees a crisp, sparse optical department. From a serial of such optical sections taken at different depths and stored in a computer, it is easy to reconstruct a three-dimensional image. The methods do for the microscopist what the CT scanner does (by different means) for the radiologist investigating a human being body: both machines requite detailed sectional views of the interior of an intact structure.
The computational approach is often called image deconvolution. To understand how it works, call back how the wave nature of lite means that the microscope lens organisation gives a small-scale blurred disc every bit the prototype of a point light source, with increased blurring if the point source lies above or below the focal aeroplane. This blurred paradigm of a bespeak source is called the bespeak spread function. An image of a complex object can so be thought of equally being built up by replacing each signal of the specimen by a respective blurred disc, resulting in an paradigm that is blurred overall. For deconvolution, we starting time obtain a serial of (blurred) images, focusing the microscope in turn on a series of focal planes—in effect, a blurred three-dimensional image. The stack of images is then processed past reckoner to remove every bit much of the blur as possible. Essentially the computer plan uses the microscope'due south signal spread function to make up one's mind what the consequence of the blurring would take been on the paradigm, and then applies an equivalent "deblurring" (deconvolution), turning the blurred three-dimensional image into a series of clean optical sections. The computation required is quite complex, and used to exist a serious limitation. Notwithstanding, with faster and cheaper computers, the paradigm deconvolution method is gaining in ability and popularity. An example is shown in Figure nine-17.

Effigy 9-17
Image deconvolution. (A) A low-cal micrograph of the large polytene chromosomes from Drosophila, stained with a fluorescent Dna-binding dye. (B) The same field of view after prototype deconvolution clearly reveals the banding pattern on the chromosomes. Each (more...)
The Confocal Microscope Produces Optical Sections by Excluding Out-of-Focus Low-cal
The confocal microscope achieves a result similar to that of deconvolution, only does so by manipulation of the lite earlier it is measured; thus it is an analog technique rather than a digital one. The optical details of the confocal microscope are complex, only the bones idea is uncomplicated, as illustrated in Figure nine-18.
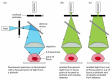
Figure 9-18
The confocal fluorescence microscope. This simplified diagram shows that the bones arrangement of optical components is similar to that of the standard fluorescence microscope shown in Figure 9-12, except that a laser is used to illuminate a small pinhole (more...)
The microscope is generally used with fluorescence eyes (see Figure 9-12), but instead of illuminating the whole specimen at once, in the usual way, the optical system at any instant focuses a spot of light onto a single betoken at a specific depth in the specimen. A very brilliant source of pinpoint illumination is required; this is normally supplied by a light amplification by stimulated emission of radiation whose light has been passed through a pinhole. The fluorescence emitted from the illuminated cloth is collected and brought to an prototype at a suitable light detector. A pinhole aperture is placed in front end of the detector, at a position that is confocal with the illuminating pinhole—that is, precisely where the rays emitted from the illuminated point in the specimen come to a focus. Thus, the low-cal from this signal in the specimen converges on this aperture and enters the detector.
By contrast, the lite from regions out of the plane of focus of the spotlight is as well out of focus at the pinhole aperture and is therefore largely excluded from the detector (Effigy nine-xix). To build up a 2-dimensional image, data from each betoken in the plane of focus are collected sequentially by scanning across the field in a raster pattern (as on a goggle box screen) and are displayed on a video screen. Although not shown in Figure 9-18, the scanning is usually washed past deflecting the beam with an oscillating mirror placed between the dichroic mirror and the objective lens in such a way that the illuminating spotlight and the confocal pinhole at the detector remain strictly in register.
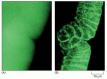
Figure ix-nineteen
Conventional and confocal fluorescence microscopy compared. These ii micrographs are of the aforementioned intact gastrula-stage Drosophila embryo that has been stained with a fluorescent probe for actin filaments. (A) The conventional, unprocessed paradigm is blurred (more than...)
The confocal microscope has been used to resolve the structure of numerous circuitous three-dimensional objects (Figure 9-20), including the networks of cytoskeletal fibers in the cytoplasm and the arrangements of chromosomes and genes in the nucleus.

Figure nine-20
Three-dimensional reconstruction from confocal microscope images. (A) Pollen grains, in this instance from a passion flower, accept a complex sculptured prison cell wall that contains fluorescent compounds. Images obtained at different depths through the grain, using (more than...)
The relative merits of deconvolution methods and confocal microscopy for three-dimensional optical microscopy are still the subject area of debate. Confocal microscopes are generally easier to utilize than deconvolution systems and the final optical sections can be seen apace. On the other paw, modernistic, cooled CCD (accuse-coupled device) cameras used for deconvolution systems are extremely efficient at collecting small amounts of lite, and they can exist used to make detailed three-dimensional images from specimens that are too weakly stained or likewise easily damaged by bright low-cal for confocal microscopy.
The Electron Microscope Resolves the Fine Structure of the Jail cell
The relationship between the limit of resolution and the wavelength of the illuminating radiation (see Figure 9-6) holds true for any form of radiation, whether information technology is a beam of lite or a axle of electrons. With electrons, even so, the limit of resolution can be fabricated very small. The wavelength of an electron decreases as its velocity increases. In an electron microscope with an accelerating voltage of 100,000 Five, the wavelength of an electron is 0.004 nm. In theory the resolution of such a microscope should exist about 0.002 nm, which is 10,000 times that of the light microscope. Because the aberrations of an electron lens are considerably harder to correct than those of a glass lens, withal, the practical resolving power of most modernistic electron microscopes is, at best, 0.1 nm (1 Å) (Effigy 9-21). This is considering only the very heart of the electron lenses can be used, and the effective numerical aperture is tiny. Furthermore, problems of specimen preparation, dissimilarity, and radiation damage take generally limited the normal effective resolution for biological objects to ii nm (20 Å). This is even so about 100 times better than the resolution of the low-cal microscope. Moreover, in contempo years, the performance of electron microscopes has been improved by the development of electron illumination sources called field emission guns. These very vivid and coherent sources tin essentially ameliorate the resolution achieved. The major landmarks in the evolution of electron microscopy are listed in Table 9-2.

Effigy 9-21
The limit of resolution of the electron microscope. This manual electron micrograph of a sparse layer of gold shows the private files of atoms in the crystal as bright spots. The distance between adjacent files of gold atoms is most 0.two nm (2 (more...)
Table ix-ii
Major Events in the Evolution of the Electron Microscope and Its Application to Prison cell Biological science.
In overall pattern the transmission electron microscope (TEM) is similar to a light microscope, although information technology is much larger and upside downwards (Figure ix-22). The source of illumination is a filament or cathode that emits electrons at the top of a cylindrical column nearly 2 g high. Since electrons are scattered by collisions with air molecules, air must first be pumped out of the column to create a vacuum. The electrons are then accelerated from the filament by a nearby anode and allowed to pass through a tiny hole to grade an electron beam that travels downwardly the column. Magnetic coils placed at intervals along the column focus the electron beam, just as glass lenses focus the light in a light microscope. The specimen is put into the vacuum, through an airlock, into the path of the electron beam. As in light microscopy, the specimen is usually stained—in this case, with electron-dense material, as nosotros run across in the adjacent section. Some of the electrons passing through the specimen are scattered past structures stained with the electron-dense material; the remainder are focused to form an prototype, in a way analogous to the fashion an epitome is formed in a light microscope—either on a photographic plate or on a phosphorescent screen. Because the scattered electrons are lost from the beam, the dumbo regions of the specimen evidence up in the prototype as areas of reduced electron flux, which look night.

Figure 9-22
The primary features of a light microscope and a transmission electron microscope. These drawings emphasize the similarities of overall pattern. Whereas the lenses in the low-cal microscope are fabricated of glass, those in the electron microscope are magnetic (more...)
Biological Specimens Crave Special Preparation for the Electron Microscope
In the early on days of its application to biological materials, the electron microscope revealed many previously unimagined structures in cells. But earlier these discoveries could be made, electron microscopists had to develop new procedures for embedding, cutting, and staining tissues.
Since the specimen is exposed to a very high vacuum in the electron microscope, there is no possibility of viewing it in the living, moisture country. Tissues are usually preserved past fixation—first with glutaraldehyde, which covalently cross-links protein molecules to their neighbors, and so with osmium tetroxide, which binds to and stabilizes lipid bilayers likewise equally proteins (Figure 9-23). Because electrons have very express penetrating power, the fixed tissues normally have to exist cut into extremely thin sections (l–100 nm thick, near 1/200 the thickness of a single cell) before they are viewed. This is accomplished by dehydrating the specimen and permeating it with a monomeric resin that polymerizes to course a solid block of plastic; the block is and then cutting with a fine drinking glass or diamond pocketknife on a special microtome. These sparse sections, free of water and other volatile solvents, are placed on a pocket-sized circular metallic grid for viewing in the microscope (Effigy 9-24).

Figure 9-23
Two common chemical fixatives used for electron microscopy. The 2 reactive aldehyde groups of glutaraldehyde enable information technology to cross-link diverse types of molecules, forming covalent bonds between them. Osmium tetroxide is reduced by many organic compounds (more...)
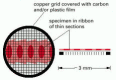
Figure 9-24
The copper filigree that supports the thin sections of a specimen in a TEM.
The steps required to set up biological material for viewing in the electron microscope accept challenged electron microscopists from the showtime. How tin can we be sure that the image of the stock-still, dehydrated, resin-embedded specimen finally seen bears whatsoever relation to the frail aqueous biological system that was originally present in the living cell? The all-time current approaches to this problem depend on rapid freezing. If an aqueous system is cooled fast enough to a depression enough temperature, the water and other components in information technology do not have time to rearrange themselves or crystallize into ice. Instead, the water is supercooled into a rigid but noncrystalline land—a "glass"—called vitreous ice. This state can exist accomplished past slamming the specimen onto a polished copper block cooled past liquid helium, past plunging it into or spraying it with a jet of a coolant such as liquid propane, or by cooling it at high force per unit area.
Some frozen specimens can be examined directly in the electron microscope using a special, cooled specimen holder. In other cases the frozen block can be fractured to reveal interior surfaces, or the surrounding water ice can exist sublimed away to betrayal external surfaces. All the same, we often want to examine thin sections, and to have them stained to yield adequate dissimilarity in the electron microscope paradigm (discussed further below). A compromise is therefore to rapid-freeze the tissue, so replace the h2o, maintained in the vitreous (glassy) state, by organic solvents, and finally embed the tissue in plastic resin, cut sections, and stain. Although technically still difficult, this approach stabilizes and preserves the tissue in a condition very close to its original living land.
Contrast in the electron microscope depends on the atomic number of the atoms in the specimen: the higher the atomic number, the more electrons are scattered and the greater the contrast. Biological tissues are composed of atoms of very low atomic number (mainly carbon, oxygen, nitrogen, and hydrogen). To make them visible, they are usually impregnated (earlier or after sectioning) with the salts of heavy metals such every bit uranium and lead. Unlike cellular constituents are revealed with various degrees of dissimilarity co-ordinate to their degree of impregnation, or "staining," with these salts. Lipids, for example, tend to stain darkly later on osmium fixation, revealing the location of jail cell membranes (Figure 9-25).
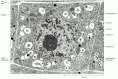
Figure 9-25
A root-tip jail cell stained with osmium and other heavy metal ions. The cell wall, nucleus, vacuoles, mitochondria, endoplasmic reticulum, Golgi appliance, and ribosomes are hands visible in this transmission electron micrograph. (Courtesy of Brian Gunning.) (more than...)
Specific Macromolecules Tin Be Localized by Immunogold Electron Microscopy
Nosotros accept seen how antibodies can be used in conjunction with fluorescence microscopy to localize specific macromolecules. An coordinating method—immunogold electron microscopy—can be used in the electron microscope. The usual procedure is to incubate a thin department with a specific primary antibody, then with a secondary antibody to which a colloidal gold particle has been attached. The gold particle is electron-dense and can be seen as a black dot in the electron microscope (Effigy 9-26).
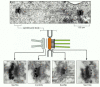
Figure 9-26
Localizing proteins in the electron microscope. Immunogold electron microscopy is used here to localize four unlike protein components to item locations within the spindle pole body of yeast. At the top is a thin section of a yeast mitotic spindle (more...)
Thin sections frequently fail to convey the 3-dimensional arrangement of cellular components in the TEM and tin be very misleading: a linear structure such as a microtubule may appear in section as a pointlike object, for case, and a section through protruding parts of a unmarried irregularly shaped solid body may requite the appearance of two or more split up objects. The third dimension tin can exist reconstructed from serial sections (Figure 9-27), but this is still a lengthy and tedious process.

Figure nine-27
A 3-dimensional reconstruction from serial sections. Unmarried thin sections sometimes requite misleading impressions. In this example, most sections through a cell containing a branched mitochondrion seem to incorporate two or three split mitochondria. (more...)
Even thin sections, however, accept a significant depth compared to the resolution of the electron microscope, and so they tin also be misleading in an opposite style. The optical design of the electron microscope—the very pocket-size aperture used—produces a large depth of field, so the image seen corresponds to a superimposition (a project) of the structures at unlike depths. A farther complication for immunogold labeling is that the antibodies and colloidal gilded particles do non penetrate into the resin used for embedding; therefore, they only notice antigens right at the surface of the department. This means that first, the sensitivity of detection is low, since antigen molecules present in the deeper parts of the section are non detected, and second, one may get a false impression of which structures incorporate the antigen and which do non. A solution to this trouble is to perform the labeling earlier embedding the specimen in plastic, when the cells and tissues are still fully accessible to labeling reagents. Extremely pocket-size gold particles, about i nm in diameter, work all-time for this procedure. Such small gold particles are usually not directly visible in the concluding sections, so boosted silver or gold is nucleated around the 1 nm golden particles in a chemical procedure very much like photographic development.
Images of Surfaces Can Exist Obtained by Scanning Electron Microscopy
A scanning electron microscope (SEM) directly produces an image of the three-dimensional structure of the surface of a specimen. The SEM is usually a smaller, simpler, and cheaper device than a transmission electron microscope. Whereas the TEM uses the electrons that have passed through the specimen to class an image, the SEM uses electrons that are scattered or emitted from the specimen's surface. The specimen to exist examined is stock-still, stale, and coated with a thin layer of heavy metallic. Alternatively, information technology can be quickly frozen, and then transferred to a cooled specimen phase for direct examination in the microscope. Often an entire constitute or small animal can exist put into the microscope with very little preparation (Figure 9-28). The specimen, prepared in whatsoever of these means, is then scanned with a very narrow axle of electrons. The quantity of electrons scattered or emitted as this primary beam bombards each successive indicate of the metallic surface is measured and used to control the intensity of a second beam, which moves in synchrony with the primary beam and forms an image on a television set screen. In this style, a highly enlarged image of the surface equally a whole is congenital up (Figure 9-29).

Figure nine-28
A developing wheat flower, or spike. This delicate bloom spike was rapidly frozen, coated with a thin metallic picture, and examined in the frozen state in a SEM. This micrograph, which is at a low magnification, demonstrates the large depth of focus of the (more than...)
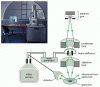
Figure 9-29
The scanning electron microscope. In a SEM, the specimen is scanned by a axle of electrons brought to a focus on the specimen by the electromagnetic coils that human activity equally lenses. The quantity of electrons scattered or emitted as the beam bombards each successive (more than...)
The SEM technique provides slap-up depth of field; moreover, since the amount of electron handful depends on the bending of the surface relative to the beam, the paradigm has highlights and shadows that give it a three-dimensional advent (Figures 9-28 and 9-thirty). But surface features can exist examined, notwithstanding, and in most forms of SEM, the resolution attainable is not very high (about 10 nm, with an effective magnification of up to xx,000 times). As a upshot, the technique is unremarkably used to study whole cells and tissues rather than subcellular organelles. Very high-resolution SEMs have, however, been recently developed with a bright coherent-field emission gun equally the electron source. This type of SEM tin produce images that rival TEM images in resolution (Figure 9-31).

Effigy ix-30
Scanning electron microscopy. (A) A scanning electron micrograph of the stereocilia projecting from a hair jail cell in the inner ear of a bullfrog. For comparing, the aforementioned construction is shown past (B) differential-interference-dissimilarity light microscopy and (more than...)
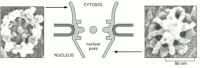
Effigy 9-31
The nuclear pore. Apace frozen nuclear envelopes were imaged in a loftier-resolution SEM, equipped with a field emission gun as the source of electrons. These views of each side of a nuclear pore correspond the limit of resolution of the SEM, and should (more...)
Metal Shadowing Allows Surface Features to Exist Examined at High Resolution past Manual Electron Microscopy
The TEM tin also be used to written report the surface of a specimen—and generally at a college resolution than in the SEM—in such a way that individual macromolecules tin exist seen. As in scanning electron microscopy, a thin motion-picture show of a heavy metal such as platinum is evaporated onto the dried specimen. The metal is sprayed from an oblique angle and so equally to deposit a coating that is thicker in some places than others—a process known as shadowing because a shadow effect is created that gives the image a 3-dimensional appearance.
Some specimens coated in this way are thin enough or small enough for the electron beam to penetrate them directly. This is the example for individual molecules, viruses, and cell walls—all of which tin can be dried down, before shadowing, onto a flat supporting film made of a material that is relatively transparent to electrons, such as carbon or plastic. For thicker specimens, the organic material of the prison cell must be dissolved away after shadowing then that only the thin metal replica of the surface of the specimen is left. The replica is reinforced with a film of carbon so it can be placed on a filigree and examined in the transmission electron microscope in the ordinary way (Effigy 9-32).

Figure 9-32
The preparation of a metallic-shadowed replica of the surface of a specimen. Note that the thickness of the metallic reflects the surface contours of the original specimen.
Freeze-Fracture and Freeze-Etch Electron Microscopy Provide Views of Surfaces Inside the Cell
Freeze-fracture electron microscopy provides a way of visualizing the interior of jail cell membranes. Cells are frozen (equally described above) and then the frozen cake is cracked with a knife bract. The fracture plane often passes through the hydrophobic middle of lipid bilayers, thereby exposing the interior of cell membranes. The resulting fracture faces are shadowed with platinum, the organic cloth is dissolved abroad, and the replicas are floated off and viewed in the electron microscope (come across Figure ix-32). Such replicas are studded with small bumps, called intramembrane particles, which represent large transmembrane proteins. The technique provides a convenient and dramatic manner to visualize the distribution of such proteins in the plane of a membrane (Figure 9-33).

Figure 9-33
The thylakoid membranes from the chloroplast of a institute cell. In this freeze-fracture electron micrograph, the thylakoid membranes, which perform photosynthesis, are stacked up in multiple layers (see Figure xiv-34). The airplane of the fracture has moved (more...)
Some other related replica method is freeze-etch electron microscopy, which can be used to examine either the exterior or interior of cells. In this technique, the frozen block is cracked with a pocketknife blade as described above. Just now the ice level is lowered around the cells (and to a lesser extent within the cells) by the sublimation of ice in a vacuum as the temperature is raised—a procedure called freeze-drying. The parts of the cell exposed by this etching process are so shadowed as earlier to make a platinum replica. This technique exposes structures in the interior of the prison cell and tin can reveal their three-dimensional organization with exceptional clarity (Figure ix-34).

Effigy nine-34
A regular array of poly peptide filaments in an insect muscle. To obtain this image, the muscle cells were speedily frozen to liquid helium temperature, fractured through the cytoplasm, and subjected to deep etching. A metal-adumbral replica was so prepared (more than...)
Negative Staining and Cryoelectron Microscopy Allow Macromolecules to Be Viewed at High Resolution
Although isolated macromolecules, such every bit Deoxyribonucleic acid or large proteins, tin can be visualized readily in the electron microscope if they are adumbral with a heavy metal to provide contrast, effectively detail can be seen past using negative staining. In this technique, the molecules, supported on a thin film of carbon, are washed with a concentrated solution of a heavy-metallic common salt such as uranyl acetate. After the sample has dried, a very thin flick of metal salt covers the carbon film everywhere except where it has been excluded past the presence of an adsorbed macromolecule. Because the macromolecule allows electrons to pass much more readily than does the surrounding heavy-metallic stain, a reversed or negative prototype of the molecule is created. Negative staining is especially useful for viewing large macromolecular aggregates such as viruses or ribosomes, and for seeing the subunit construction of protein filaments (Effigy nine-35).

Effigy 9-35
Negatively stained actin filaments. In this transmission electron micrograph, each filament is about viii nm in diameter and is seen, on close inspection, to be composed of a helical chain of globular actin molecules. (Courtesy of Roger Craig.)
Shadowing and negative staining can provide loftier-contrast surface views of small macromolecular assemblies, but both techniques are limited in resolution past the size of the smallest metal particles in the shadow or stain used. Recent methods provide an alternative that has allowed even the interior features of three-dimensional structures such as viruses to be visualized straight at high resolution. In this technique, called cryoelectron microscopy, rapid freezing to form vitreous ice is again the key. A very thin (about 100 nm) film of an aqueous pause of virus or purified macromolecular complex is prepared on a microscope grid. The specimen is then rapidly frozen by plunging it into a coolant. A special sample holder is used to keep this hydrated specimen at -160°C in the vacuum of the microscope, where it can exist viewed directly without fixation, staining, or drying. Unlike negative staining, in which what is seen is the envelope of stain exclusion around the particle, hydrated cryoelectron microscopy produces an epitome from the macromolecular structure itself. However, to extract the maximum corporeality of structural information, special image-processing techniques must be used, as nosotros depict next.
Multiple Images Tin can Be Combined to Increment Resolution
Whatsoever epitome, whether produced by an electron microscope or by an optical microscope, is fabricated past particles—electrons or photons—striking a detector of some sort. Merely these particles are governed by breakthrough mechanics, so the numbers reaching the detector are predictable only in a statistical sense. In the limit of very large numbers of particles, the distribution at the detector is accurately determined past the imaged specimen. Nevertheless, with smaller numbers of particles, this underlying construction in the image is obscured by the statistical fluctuations in the numbers of particles detected in each region. Random variability that confuses the underlying image of the specimen itself is referred to as racket. Dissonance is a particularly severe problem for electron microscopy of unstained macromolecules, just it is also important in low-cal microscopy at depression lite levels. A protein molecule can tolerate a dose of just a few tens of electrons per square nanometer without damage, and this dose is orders of magnitude below what is needed to define an image at diminutive resolution.
The solution is to obtain images of many identical molecules—perhaps tens of thousands of individual images—and combine them to produce an averaged image, revealing structural details that were subconscious by the noise in the original images. Before the individual images tin be combined, all the same, they must be aligned with each other. Sometimes it is possible to induce proteins and complexes to form crystalline arrays, in which each molecule is held in the same orientation in a regular lattice. In this example, the alignment problem is easily solved, and several poly peptide structures have been determined at atomic resolution past this type of electron crystallography. In principle, however, crystalline arrays are non absolutely required. With the help of a computer, the images of randomly distributed molecules tin be processed and combined to yield high-resolution reconstructions, as nosotros now explain.
Views from Different Directions Tin can Be Combined to Requite 3-dimensional Reconstructions
The detectors used to record images from electron microscopes produce 2-dimensional pictures. Because of the big depth of field of the microscope, all the parts of the three-dimensional specimen are in focus, and the resulting image is a projection of the structure along the viewing management. The lost data in the 3rd dimension tin be recovered if we have views of the same specimen from many different directions. The computational methods for this technique were worked out in the 1960s, and they are widely used in medical computed tomography (CT) scans. In a CT browse, the imaging equipment is moved around the patient to generate the dissimilar views. In electron-microscope (EM) tomography, the specimen holder is tilted in the microscope, which achieves the same issue. In this manner, 1 tin arrive at a iii-dimensional reconstruction, in a called standard orientation, by combining a set of views of many identical molecules in the microscope'southward field of view. Each view volition be individually very noisy, just past combining them in three dimensions and taking an boilerplate, the noise can be largely eliminated, yielding a articulate view of the molecular structure.
EM tomography is now widely applied for determining both molecular structures, using either crystalline or noncrystalline specimens, and larger objects such every bit thin sections of cells and organelles. It is a especially successful technique for structures that accept some intrinsic symmetry, such as helical or icosahedral viruses, because it makes the task of alignment easier and more accurate. Figure 9-36 shows the structure of an icosahedral virus that has been determined at high resolution by the combination of many particles and multiple views, and Effigy ix-37 shows the construction of a ribosome adamant in the aforementioned way.

Effigy nine-36
EM tomography. Spherical poly peptide shells of the hepatitis B virus are preserved in a thin film of ice (A) and imaged in the transmission electron microscope. Thousands of individual particles were combined by EM tomography to produce the three-dimensional (more...)
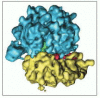
Figure 9-37
The three-dimensional structure of the 70S ribosome from E. coli adamant by EM tomography. The small subunit is colored xanthous, the large subunit blue. The overall resolution is 11.2 Å. (From I.S. Gabashvili et al., Cell 100: 537–549, (more than...)
With crystalline arrays, a resolution of 0.3 nm has been accomplished by electron microscopy—enough to begin to run into the internal atomic arrangements in a protein and to rival x-ray crystallography in resolution. With unmarried-particle reconstruction, the limit at the moment is about 0.eight nm, plenty to place protein subunits and domains, and limited protein secondary construction. Although electron microscopy is unlikely to supersede x-ray crystallography (discussed in Chapter 8) every bit a method for macromolecular structure determination, information technology has some very clear advantages. Get-go, it does not absolutely require crystalline specimens. Second, it can bargain with extremely big complexes—structures that may be likewise large or also variable to crystallize satisfactorily. Electron microscopy provides a bridge between the scale of the unmarried molecule and that of the whole prison cell.
Summary
Many calorie-free-microscope techniques are available for observing cells. Cells that have been stock-still and stained can be studied in a conventional light microscope, while antibodies coupled to fluorescent dyes can exist used to locate specific molecules in cells in a fluorescence microscope. Living cells can exist seen with stage-dissimilarity, differential-interference-contrast, night-field, or vivid-field microscopes. All forms of light microscopy are facilitated by electronic image-processing techniques, which enhance sensitivity and refine the image. Confocal microscopy and image deconvolution both provide thin optical sections and tin can exist used to reconstruct iii-dimensional images.
Determining the detailed structure of the membranes and organelles in cells requires the higher resolution accessible in a transmission electron microscope. Specific macromolecules tin be localized with colloidal gilt linked to antibodies. Three-dimensional views of the surfaces of cells and tissues are obtained by scanning electron microscopy. The shapes of isolated macromolecules that accept been shadowed with a heavy metallic or outlined by negative staining can as well be readily determined by electron microscopy. Using computational methods, multiple images and views from dissimilar directions are combined to produce detailed reconstructions of macromolecules and molecular complexes through a technique known as electron-microscope tomography.

What Are The Sizes Of A Molecule, Bacteria, Animal Cell, Virus, And Plant Cell?,
Source: https://www.ncbi.nlm.nih.gov/books/NBK26880/
Posted by: jordanmandes.blogspot.com
0 Response to "What Are The Sizes Of A Molecule, Bacteria, Animal Cell, Virus, And Plant Cell?"
Post a Comment